The ancestor of SARS-CoV-2’s Wuhan strain was circulating in late October 2019

The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in a case from Wuhan, China, in December 2021, and subsequently became the cause of the coronavirus disease 2019 (COVID-19) pandemic that is ravaging the world today. A new study in the journal Molecular Biology and Evolution tracks its variants all over the world since the onset of the pandemic.
Genomic sequencing has occurred using hundreds of thousands of viral genomic samples. The researchers used the best of these sequences to reveal how the virus has mutated and changed in different periods and regions of the pandemic.
.jpg)
Using a new approach
Conventional methods have not produced a reliable history of the emergence of this virus for several reasons. These include the numerous widespread sequencing errors in the available sequences, small degree of sequence divergence, and the fact that there are few sites that help to understand the descent of the virus.
Especially remarkable is the fact that all known early SARS-CoV-2 genomes from humans (up to January 2020) vary by less than 30 bases. Conversely, the most closely related non-human coronaviruses differ by over a thousand bases.
The importance of this is that “Without a reliable root of the SARS‐CoV‐2 phylogeny, the most recent ancestor sequence cannot be accurately reconstructed, and it is not possible to assess the genetic diversity of SARS‐CoV‐2 that existed at the time of its first outbreak.”
Moreover, the distance of the Wuhan samples from the progenitor will remain unknown, as will the direction and order of the first mutations that gave rise to the various strains and lineages of SARS-CoV-2.
They used computational methods originally designed to find how mutations occurred within tumor cells in a single patient. The approach used is called a mutation order approach (MOA) and can provide a direct picture of the ancestral variants and mutations in order of time.
What were the findings?
MOA was employed on two sets of SARS‐CoV‐2 genomes, comprising almost 30,000 and 68,000 genomes, respectively, on two days three months apart. By tracing the mutational trail, inferred from the second genome set, they were able to understand how the virus is undergoing changes in different regions and at different times. They were able to track back to the most recent common ancestor (MRCA) of SARS-CoV-2.
Common ancestor
This progenitor viral genome has three bases that differ from the Wuhan strains. The researchers think that both the Wuhan and other of the earliest genomes to be sampled were actually variants of the progenitor coronavirus (CoV), which diverged into ν and α lineages.
Diversity pre-existing the earliest case
The Wuhan strain underwent three consecutive mutations, α1, α2, and α3, but these are not found in the closely related CoVs, all of which have the same base at these three positions. The ν variants of the progenitor CoV do not show the other 47 variants at these positions, making them unlikely to be the ancestral lineage for the Wuhan-1 virus or other early samples. The first ν mutant was picked up almost two months after the Wuhan-1 strain.
There were multiple occurrences of the progenitor CoV, both in China and the USA, from January 2020 onwards. Synonymous progenitor CoV samples were found in many other samples collected within two weeks of the Wuhan-1 strain.
While these were mostly Chinese and Asian (almost 90/130), they were found in all continents sampled and persisted up to April 2020 in Europe.
These findings suggest that the progenitor CoV was already spreading extensively before and after the first official reports of the emergence of a novel coronavirus in China. In other words, the Wuhan-1 strain is unlikely to be the original SARS-CoV-2 ancestor from which all currently circulating strains are derived.
This is in contrast to earlier studies, probably because this analysis uses more samples from a global database, and thus identifies the very early ν lineage, which is nonetheless not the MRCA. The latter is thought to have preceded the Wuhan variant by 6-8 weeks, that is, late October 2019.
In fact, Italian scientists found a spike protein fragment from SARS-CoV-2 in Italy in early December, that exactly resembled the Wuhan-1 genome.
The analysis of the second set of genomes showed the same pattern leading to the same conclusions. Two new mutations were identified belonging to the ζ and η groups from the middle of March 2020.
Viral genomic fingerprints allow tracking over time and region
The mutational history led to a collection of genetic fingerprints extending from the progenitor CoV to the current strain. Each is named for the major variants included.
Both this progenitor genome and its branches have since led to an array of lineages or strains, some of which (e.g., D614G) have rapidly ascended to global or regional dominance in a very short time.
The North American strains have all belonged to the same lineages for the major part of the pandemic period. These were mostly αβ along with its mutant (αβγδ), which has remained dominant since April 2020.
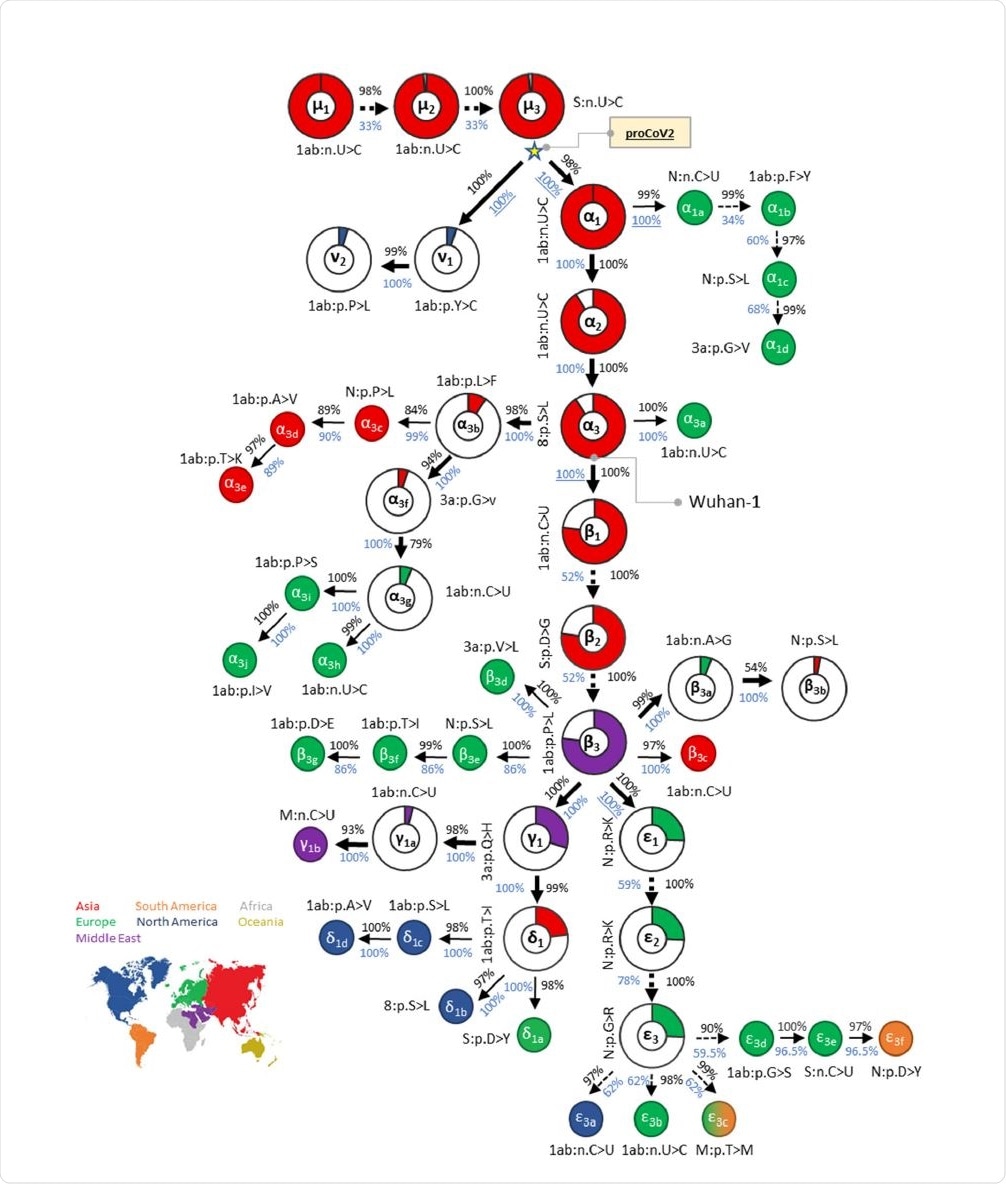
In Asia and Europe, the rate of change in dominant strains is high, converging to αβε by July to August 2020, and later αβεη. These then shifted to αβζ, beginning at three weeks from the first ε variant samples. Many strains continue to circulate at high frequencies in both Asia and North America.
The South African variant has the αβγδ genetic fingerprint, and the UK variant the αβε genetic fingerprint. Both have the N501Y spike mutation, and both show identical properties of higher infectiousness. At the time of the study, the αβζ fingerprint was dominant, while today, probably the UK variant has taken over.
What are the conclusions?
The researchers have identified the MRCA for the SARS-CoV-2 variants circulating today, which is probably not the Wuhan-1 virus but its progenitor. This implies “that none of the earliest patients represent the index case or gave rise to all the human infections.”
The MOA approach used here yielded the progenitor CoV genome, which gives a better rooted phylogenetic tree, mutational order and divergent mutations in genomic sequences. The approach will be relevant to any such pathogenic outbreaks, even with larger samples which may, in fact, yield more accurate results.
Its continued application to SARS‐CoV‐2 genomes and other pathogen outbreaks will produce their ancestral genomes and their spatiotemporal dynamics, improving our understanding of the past, current, and future evolution of pathogens and associated diseases.”
The researchers have set up a dashboard that will be constantly updated with newly emerging mutations and will reflect the trends of viral spread over time and by region. In addition, a simple tool is provided to classify any given genome by key mutations (http://sars2evo.datamonkey.org/).
- Kumar, S. et al. (2021). An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic. Molecular Biology and Evolution. https://doi.org/10.1093/molbev/msab118, https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msab118/6257226
Posted in: Medical Science News | Medical Research News | Disease/Infection News | Healthcare News
Tags: Bases, Coronavirus, Coronavirus Disease COVID-19, Evolution, Genetic, Genome, Genomic, Genomic Sequencing, Molecular Biology, Mutation, Pandemic, Pathogen, Phylogeny, Protein, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, Tumor, Virus

Written by
Dr. Liji Thomas
Dr. Liji Thomas is an OB-GYN, who graduated from the Government Medical College, University of Calicut, Kerala, in 2001. Liji practiced as a full-time consultant in obstetrics/gynecology in a private hospital for a few years following her graduation. She has counseled hundreds of patients facing issues from pregnancy-related problems and infertility, and has been in charge of over 2,000 deliveries, striving always to achieve a normal delivery rather than operative.
Source: Read Full Article
Related posts:
 Ivermectin may not be the ‘silver bullet’ antiviral against COVID-19
Ivermectin may not be the ‘silver bullet’ antiviral against COVID-19
 Does climate play a large role in SARS-CoV-2 transmission?
Does climate play a large role in SARS-CoV-2 transmission?
 GSK-3 inhibitors show promise in treating coronavirus infections
GSK-3 inhibitors show promise in treating coronavirus infections
 How effective has rapid COVID-19 vaccination been in Israel?
How effective has rapid COVID-19 vaccination been in Israel?
 Researchers develop new rapid and highly sensitive SAR-CoV-2 RNA detection test
Researchers develop new rapid and highly sensitive SAR-CoV-2 RNA detection test